Distrofia miotónica
Distrofia miotónica (português europeu) ou distrofia miotônica (português brasileiro) é uma doença genética crónica que afeta a função muscular.[1] Os sintomas mais evidentes são fraqueza e atrofia muscular progressivas.[1] Em muitos casos os músculos contraem-se e são incapazes de relaxar.[1] Entre outros possíveis sintomas estão cataratas, deficiência intelectual e problemas na condução elétrica do coração.[1][2] Em homens, pode ocorrer calvície precoce e infertilidade.[1]
| Distrofia miotónica | |
|---|---|
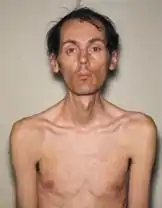 Distrofia miotónica | |
| Sinónimos | Miotonia atrófica,[1] miotonia distrófica[1] doença de Steinert, distrofia muscular de Steinert |
| Especialidade | Genética médica, pediatria |
| Sintomas | Atrofia muscular, fraqueza, músculos que se contraem e são incapazes de relaxar[1] |
| Complicações | Cataratas, deficiência intelectual, problemas na condução elétrica do coração[1][2] |
| Início habitual | 20 a 40 anos de idade[1] |
| Duração | Crónica[1] |
| Tipos | Tipo 1, tipo 2[1] |
| Causas | Genéticas (autossómica dominante)[1] |
| Método de diagnóstico | Exames genéticos[2] |
| Tratamento | Ortóteses, cadeira de rodas, pacemaker, ventilação com pressão positiva não invasiva[2] |
| Medicação | Mexiletina, carbamazepina, antidepressivos tricíclicos, anti-inflamatórios não esteroides[2] |
| Frequência | >1 em 8 000 pessoas[1] |
| Classificação e recursos externos | |
| CID-10 | G71.11, G71.1 |
| CID-9 | 359.21 |
| CID-11 | 192087511 |
| OMIM | 160900 |
| DiseasesDB | 8739 |
| MeSH | D009223 |
A distrofia miotónica é uma doença genética autossómica dominante que geralmente é herdada de um dos progenitores.[1] Existem dois tipos principais: o tipo 1 (DM1), causado por mutações no gene DMPK, e o tipo 2 (DM2), causado por mutações no gene CNBP.[1] A doença geralmente agrava-se a cada geração.[1] O tipo DM1 pode ser evidente ao nascer.[1] O tipo DM2 é geralmente menos grave.[1] A distrofia miotónica é um tipo de distrofia muscular.[1] O diagnóstico é confirmado com exames genéticos.[2]
Não existe cura.[3] O tratamento pode incluir a utilização de ortóteses, cadeira de rodas, pacemaker e ventilação com pressão positiva não invasiva.[2] Em alguns casos pode ser útil a administração de mexiletina ou carbamazepina.[2] A ocorrência de dor pode ser tratada com antidepressivos tricíclicos e anti-inflamatórios não esteroides.[2]
A distrofia miotónica afeta mais de 1 em cada 8000 pessoas em todo o mundo.[1] Embora a doença se possa começar a manifestar em qualquer idade, o início mais comum é entre os 20 e 40 anos de idade.[1] A distrofia miotónica é a forma mais comum de distrofia muscular com início em idade adulta.[1] A doença foi descrita pela primeira vez em 1909 e em 1992 foi determinada a causa subjacente do tipo 1.[2]
Referências
- «myotonic dystrophy». GHR. 11 de outubro de 2016. Consultado em 16 de outubro de 2016. Cópia arquivada em 18 de outubro de 2016
- Meola, G; Cardani, R (abril de 2015). «Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms.». Biochimica et Biophysica Acta. 1852 (4): 594–606. PMID 24882752. doi:10.1016/j.bbadis.2014.05.019
- Klein, AF; Dastidar, S; Furling, D; Chuah, MK (2015). «Therapeutic Approaches for Dominant Muscle Diseases: Highlight on Myotonic Dystrophy.». Current Gene Therapy. 15 (4): 329–37. PMID 26122101. doi:10.2174/1566523215666150630120537
Ligações externas
- Distrofia miotónica no Manual Merck